Klebsiella "Explainers"
The bacteria that comprise the genus Klebsiella are found ubiquitously in the environment (soil and water), colonising plants, animals, and humans. For this reason, different scientific disciplines are interested in studying the many species in the Klebsiella genus. Certain species are if critical importance due to their propensity to cause human diseases, many of which are resistant to commonly used antibiotics. As such, scientists and clinicians have been interested in understanding more about the Klebsiella genus, including: accurately identifying their species; and categorising other characteristics, for example virulence determinants or antimicrobial resistance (AMR) genes.
Klebsiella species (spp.) have become increasingly problematic amongst human populations in recent years, as antibiotic use amongst humans and animals has expanded. Now some Klebsiella spp. have developed resistance to the entire antimicrobial arsenal used in humans. The World Health Organisation lists Klebsiella amongst its top 3 priority pathogens. Multi-drug resistant organisms can cause hospital outbreaks and pose a significant public health threat.
Challenges with determining Klebsiella species
The Klebsiella genus consists of Gram negative, oxidase negative bacilli. Klebsiella spp. exhibit wide phenotypic and genotypic variation. There are 13 species described, of which five closely related species are within the Klebsiella pneumoniae species complex (see the phylogenetic tree of type strains). For the most well-studied species, Klebsiella pneumoniae, genomic methods have demonstrated extensive diversity, identifying many deep branching lineages with wide geographic variability in human gut carriage and disease collections.
Many different Klebsiella spp. colonise humans, animals and the environment and this exposure can result in infections occurring in healthy individuals (with hypervirulent strains) or immunocompromised hosts (opportunistic strains). It is, therefore, crucial to be able to rapidly and accurately speciate Klebsiella, which in the clinical laboratory is most frequently done using biochemical methods and increasingly, mass spectrometry (MALDI-TOF). However, Klebsiella species demonstrate overlapping phenotypes and can therefore be difficult to distinguish, especially those closely related within the Klebsiella pneumoniae species complex (Kp1-7). These are generally not well characterised using non-genomics methods, which can hinder the ability to accurately link certain species/strains to clinical/disease phenotypes. Further, the genus Raoultella is closely related to Klebsiella, but can be distinguished through biochemical and genotypic methods, and has the ability to also cause opportunistic infections.
Klebsiella species information
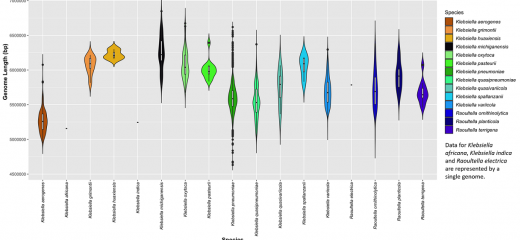
There have been 13 Klebsiella species identified as of July 8th 2020, and 4 species in the closely-related genus Raoultella.
| Species (Phylogroup) | Details |
|---|---|
| Klebsiella pneumoniae species complex | |
| Klebsiella pneumoniae (Kp1) | The most clinically important Klebsiella species. K. pneumoniae are present in the human microbiota of the gut, nose, mouth and skin. There is a wide diversity of this species carried at these sites and on occasion, these bacteria can cause invasive disease, particularly in immunocompromised hosts (elderly, hospitalised, neonates), as an opportunistic pathogen causing pneumonia, urinary tract infections, and soft tissue infections. There are also hypervirulent strains of K. pneumoniae that cause disease in otherwise healthy hosts, through the acquisition of virulence gene causing pneumonia, meningitis and liver abscesses. K. pneumoniae can carry extended spectrum beta lactamase (ESBL) and carbapenem-resistant Enterobacteriaceae (CRE) genes and share these with other organisms. These are a frequent cause of hospital outbreaks and in many parts of the world and antimicrobial resistant (AMR) Klebsiella can become untreatable with our current antibiotic arsenal. |
| K. quasipneumoniae subsp quasipneumoniae (Kp2) Subsp similioneumoniae (Kp4) |
K. quasipneumoniae were first identified amongst human blood culture isolates causing invasive disease in 2014. |
| K. quasivariicola (Kp6) | K. quasivariicola were identified from a collection of ESBL Klebsiella spp. isolates from patient samples in the United States. The isolate was not collected from invasive disease but a wound swab. |
| K.africana (Kp7) | K. africana (originally named K. africanensis) have been identified in human faecal carriage sample from Senegal and in human clinical isolates from Kenya. |
| K. variicola subsp tropica (Kp5) subsp variicola (Kp3) |
K. variicola colonise plants (rice, sugar cane, banana), insects, animals, and humans and were identified as a species separate to K. pneumoniae in 2004. The nitrogen-fixing abilities of K. variicola are beneficial in environmental settings, including its relationship with the leaf cutter ant. K. variicola can cause disease in plants (ironwood tree), animals (bovine mastitis) and humans. Their ability to produce carbapenemases and some colistin-resistant strains make this AMR/MDR gene reservoir particularly problematic. |
| Other species | |
| K. aerogenes | K. aerogenes are found as part of the human gut microbiota. They are not normally pathogenic organisms but can cause opportunistic infections. K. aerogenes have inducible resistance mechanisms which may further complicate treatment in human populations. K. aerogenes also have hydrogen-producing activity and this has been exploited commercially. |
| K. michiganensis | K. michiganensis were first isolated from the surface of a toothbrush holder in 2013, in a microbial ecology study from a household in Michigan. They have been identified in invasive disease as an opportunistic pathogen in immunocompromised and neonatal hosts harbouring ESBL and KPC genes. |
| K. pasteurii | K. pasteurii were identified in 2019 from the faecal samples from humans, cows and turtles collected in Italy. td=""> |
| K. grimontii | K. grimontii were identified in 2018 through isolates from human infections (blood cultures, superficial infections, haemorrhagic colitis) and human faecal carriage. |
| K. oxytoca | K. oxytoca are present in the human microbiota of the gut, nose, mouth and skin. They are the second most common species of Klebsiella to cause human infections, after K. pneumoniae. Infections often occur in hospital environments and are considered opportunistic, occurring in immunosuppressed hosts and causing a range of infections including pneumonia, urinary tract infections and soft tissue/wound infections. K. oxytoca are intrinsically resistant to amino- and carboxypenicillins due to the blaOXY gene and can carry ESBL and CRE genes which can also be shared with other organisms. K. oxytoca are of interest in the industry, given their nitrogen-fixing and other metabolic properties. |
| K. huaxiensis | K. huaxiensis were identified in 2018 causing a human urinary tract infection in a hospitalised patient in China. |
| K. spallanzanii | K. spallanzanii were isolated in 2019 from samples of human urine and bovine faeces collected in Italy. |
| K. indica | K. indica were isolated in 2020 from the surface of a tomato collected in a vegetable market in India. |
| Raoultella terrigena | Raoultella terrigena are naturally occurring in environmental niches including soil and water. They are rarely responsible for causing human disease. |
| Raoultella ornithinolytica | Raoultella ornithinolytica are found in aquatic environments and fish. They are increasingly recognised as a cause of human opportunistic infections with a broad spectrum including: bone, biliary tract, urinary tract, intracranial, pneumonia and bloodstream infections. |
| Raoultella planticola | Raoultella planticola are naturally occurring in environmental niches including soil and water. They are emerging as a recognised cause of invasive disease, largely in immunocompromised hosts. |
| Raoultella electrica | Raoultella electrica were isolated in 2018 from the surface of a hen’s eggs, collected from vendors in India. |

Phylogenetic tree demonstrating the 13 Klebsiella and 4 Raoultella type strains.
Defining Klebsiella type strains
We identified 17 type strain isolates from an examination of the NCBI Assembly information and cross-referenced this with the defined species for Klebsiella/Raoultella at NCBI Taxonomy and references therein (Table). The genomes for type strains of 3 Klebsiella species were re-annotated from an analysis of the literature, namely K. africana, K. spallanzanii and K. pasteurii. The NCBI Assembly information for these genomes was overruled so that the PubMLST Multi-species database could reflect the latest species definitions. Where more than one type strain for a species was identified (e.g. if there are two substrains defined), only one type strain was selected to represent the species, to simplify the species-level analysis.
| Multi-species Database ID |
Isolate Name | Species | NCBI Assembly Database Accession | NCBI BioSample Accession | Country | Year of Isolation |
|---|---|---|---|---|---|---|
| 55531 | KCTC 2190 | Klebsiella aerogenes | GCA_000215745.1 | SAMN02603581 | n/a | n/a |
| 372794 | SB5857 | Klebsiella africana | GCA_900978845.1 | SAMEA4969318 | n/a | n/a |
| 252644 | 06D021 | Klebsiella grimontii | GCA_900200035.1 | SAMEA104188450 | France | 1997 |
| 372817 | WCHKl090001 | Klebsiella huaxiensis | GCA_003261575.2 | SAMN08861555 | China | 2017 |
| 400208 | TOUT106 | Klebsiella indica | GCA_005860775.1 | SAMN11776007 | India | 2014 |
| 299619 | DSM 25444 | Klebsiella michiganensis | GCA_002925905.1 | SAMN07187491 | USA | 2010 |
| 173238 | NBRC 105695 | Klebsiella oxytoca | GCA_001598695.1 | SAMD00046899 | USA | n/a |
| 418978 | SB6412 | Klebsiella pasteurii | GCA_902158725.1 | SAMEA5610074 | Italy | 2017 |
| 70926 | DSM 30104 | Klebsiella pneumoniae | GCA_000281755.1 | SAMN02470063 | n/a | n/a |
| 173839 | 01A030 | Klebsiella quasipneumoniae | GCA_000751755.1 | GCA_000751755.1 | Austria | 1997 |
| 271266 | KPN1705 | Klebsiella quasivariicola | GCA_002269255.1 | SAMN06438648 | USA | 2014 |
| 418995 | SB6411 | Klebsiella spallanzanii | GCA_902158555.1 | SAMEA5610068 | Italy | 2017 |
| 123691 | DSM 15968 | Klebsiella variicola | GCA_000828055.2 | SAMN01174581 | Mexico | n/a |
| 401343 | DSM 102253 | Raoultella electrica | GCA_006711645.1 | SAMN12125603 | Japan | 2013 |
| 178728 | NBRC 105727 | Raoultella ornithinolytica | GCA_001598295.1 | SAMD00046738 | Japan | n/a |
| 178731 | ATCC 33531 | Raoultella planticola | GCA_000735435.1 | SAMN02743268 | n/a | n/a |
| 377940 | NCTC13038 | Raoultella terrigena | GCA_900706855.1 | SAMEA2580322 | n/a | n/a |
Genomic methods
Methods used to validate species
(1) rMLST Ribosomal Nucleotide Identity: calculated from scanning a query genome against a library of rMLST alleles for the 17 type strains using BLASTN and extracting the number of identical bases per allelic match from the BLASTN output. A global sequence identity was calculated for each type strain ribosomal ‘mini-genome’.
(2) Whole genome Average Nucleotide Identity (wgANI): calculated for each query isolate against the whole genome sequences of the 17 type strains using OrthoANI software (Lee et al., 2015).
(3) Kleborate software species scan: uses MASH software (Ondov et al., 2016) to search a genome sequence against a library of k-mer sketches derived from a library of reference genomes (Wick et al., 2018).
Kleborate Species Scan
The MinHash sketch library downloaded with the Kleborate software contains 1,034 entries for Klebsiella/Raoultella genomes out of a total of 2,620 library entries. Examination of the Kleborate library revealed parameters set to a k-mer size of 21 and 1,000 k-mers per sketch. The MASH software is used to search a query genome against the sketch library by first converting the query genome into a sketch using the MinHash algorithm and then comparing the number of k-mers in common with each sketch library entry. Kleborate parses the MASH output to identify the library entry sharing the most k-mers with the query genome and describes the match as either ‘strong’ or ‘weak’ based on the observed distance metric generated from the pairwise comparison.
What is rMLST (Ribosomal Multilocus Sequence Typing)?
rMLST is a multilocus typing system that indexes the variation of the bacterial protein-encoding ribosomal genes (Jolley et al., 2012). Gene variants (also called alleles) are numbered in order of discovery, with each unique sequence given a new allele number (Figure 1). They are stored in the rMLST sequence definition library on the PubMLST website (https://pubmlst.org/species-id). There are 53 genes/loci defined in the rMLST scheme from across the 30S and 50S subunits of the ribosome. The scheme does not include 16S RNA.

Figure 1: An example of the rplF gene (also called BACT000035), showing the allele number 2393.
The gene sequence is 534 base pairs in length. This allele is found in some Klebsiella aerogenes isolates.
A genome sequence is annotated with the rMLST alleles for each of the rMLST loci. This is an iterative process requiring the genomic DNA sequence to be scanned against the library of existing alleles using BLASTN. Matches to existing rMLST alleles are assigned to the genome, while regions of DNA that closely match existing alleles are identified, extracted and a new rMLST allele record is created in the sequence definition library. A second BLASTN scan is required to identify the region of the genome matching the newly defined allele, which is then assigned to the genome. In this iterative manner, genomes in the PubMLST Multi-species isolate database (registration required, no fee for academic use) are systematically assigned rMLST alleles (Figure 2).

Figure 2: rMLST allele designations for all 53 loci of the isolate called KCTC 2190 isolate id 55531, Klebsiella aerogenes
After assigning an allele number to each of the 53 rMLST loci, the complete scheme (Figure 2) can be used to determine unique profiles, similar to the way this is done for conventional 7 locus MLST. A complete set of rMLST alleles can be assigned as an rMLST profile or ribosomal sequence type (rST). For example, the unique combination of alleles observed for KCTC 2190 is defined as rST:44287 and annotated as Klebsiella aerogenes as KCTC 2190 is known to be the type strain for the species. Two loci were excluded from the profile (assigned a letter N in the sequence definition database) as they are known to be paralogous, with multiple versions of the gene found in the genome. These are BACT000060 (rpmE) and BACT000065 (rpmJ), which are typical of members of the Enterobacteriaceae family. If another genome is found to have this exact combination of rMLST alleles, the isolate can be assigned rST:44287 and the associated species annotation of Klebsiella aerogenes.
Phylogenetic analysis using rMLST alleles
The DNA of all 53 of the rMLST alleles for this isolate entry can be concatenated into a ‘mini-genome’ and used to rapidly compare with other isolates. Phylogenetic trees can be generated by searching for a dataset of isolates and then clicking the iTOL (Interactive Tree of Life) button in the analysis tool section at the bottom of the results page (Figure 3).

Figure 3: Screenshot of the analysis tools section present at the bottom of an isolate search results page. The iTOL (Interactive Tree of Life) button is listed in the Third Party tools section.
Clicking on the iTOL button opens up a new page of options that must be completed before the calculations are performed (Figure 4). The list of isolate identifiers has been automatically populated from the previous search page. Clicking on the Ribosomal MLST checkbox in the ‘Schemes’ section will ensure that the calculation uses all 53 rMLST loci. Paralogous loci are automatically identified and removed from the final multiple sequence alignment used for building the tree. The tree can be also be annotated with information extracted from the isolate records under the ‘iTOL datasets’ section.

Figure 4: Screenshot of the iTOL options page with 5 main parts a) list of isolate identifiers b) option to include/upload user-defined genome sequences, where you could add you own genomes of interest to compare to type strains for example c) list of loci to include in the analysis d) list of schemes to include in the analysis e) metadata to include in the annotation of the tree, isolate name and species metadata are highlighted.
The rMLST allele designations of each isolate in the list are extracted from the PubMLST isolate database and the DNA sequences for each allele are downloaded. A multiple sequence alignment (MSA) of the alleles within each individual rMLST locus is performed separately, and the resulting alignments are concatenated to give an aligned ‘mini-genome’ of each isolate. Paralogous loci are automatically identified based on the presence of multiple allele designations for a single locus per isolate. These are not included concatenated multiple sequence alignment. The phylogenetic tree based on the mini-genome MSA is calculated using FastTree. The tree and meta-data files are automatically uploaded to the iTOL website where customisation of the tree can occur using the iTOL web interface (Figure 5).

Figure 5: Phylogenetic tree of 17 type strain isolates in the Klebsiella/Raoultella genera with one entry from NCBI Assembly, Strain: 2880STDY5682802 (ID:173231) annotated as Klebsiella oxytoca (as of 8th July 2020). The tree was calculated from the concatenated alleles of 51 rMLST loci. The isolate clusters with the type strain for Raoultella planticola.
PubMLST Multi-species isolate database
The PubMLST Multi-species isolate database contains isolate data and associated genome sequences obtained from two main sources:
- NCBI Assembly database (https://www.ncbi.nlm.nih.gov/assembly)
- Genomes assembled in-house from data at the ENA Sequence Read Archive (ENA-SRA, https://www.ebi.ac.uk/ena)
The database was started in 2012 and has increased in size as genomes have become available at the NCBI Assembly database. Genomes have been assembled from ENA-SRA sequencing reads for selected species of interest to the Maiden Group, particularly for Neisseria, Campylobacter and Streptococcus-related projects. The database contains a total over 350,000 isolates with genomes across more than 11,500 different species (as of 8th July 2020).
Over time, new species are discovered and their genomes are sequenced and added to the NCBI Assembly database. In turn, these are imported into the PubMLST Multi-species database. In addition, isolates can be taxonomically re-classified as the species definitions are updated and genomic analyses show that an isolate previously thought to be species A is now species B.
Due to the relatively large number of new species recently defined for the Klebsiella genus, a review of the species annotations for all Klebsiella isolates was conducted to ensure that the PubMLST Multi-species database was consistent with the new species definitions. Raoultella isolates were also included in the analysis as they are the most closely related genus to Klebsiella.
Each Klebsiella/Raoultella isolate was annotated with rMLST allele designations (described in more detail in the Explainers: What is rMLST?) to facilitate the rapid calculation of sequence similarity between genomes. By pre-calculating the rMLST allele indexes for each rMLST locus, the time-consuming BLASTN searches are not required to be run at the point of genomic comparison.
Species identifier
Ribosomal Nucleotide Identity (RNI)
For any two genomes, the rMLST alleles can be identified and the number of identical bases between each pair of alleles for the same locus calculated from a pairwise alignment. Summing over all rMLST loci (excluding paralogous loci) and dividing by the total number of bases in all rMLST alleles of one of the genomes (usually designated as the reference genome) gives a global sequence identity.
We have developed an efficient method for scanning a query genome sequence against the rMLST allele sequences denoted by the rMLST profiles of a library of isolates. This method has been used to derive the Ribosomal Nucleotide Identity of each pairwise comparison. The number of aligned alleles and bases are also recorded.
A webserver that scans a genome sequence against the rMLST alleles of a library of Klebsiella/Raoultella type strain isolates in order to calculate the Ribosomal Nucleotide Identity of each pairwise comparison has been developed (https://pubmlst.org/species-id/klebsiella).
An example of a genomic scan using PubMLST Multi-species database entry id:87001 (Isolate name: UCI 27, Species: Klebsiella aerogenes) is shown in Figure 1. The top 5 results are shown ranked by Ribosomal Nucleotide Identity so that the most similar isolate in the library is ranked first. The pairwise comparison between the query genome and library id:55531 (Species: Klebsiella aerogenes) has an RNI value of 99.947% with the ‘mini-genome’ DNA sequence of the rMLST profile of the library entry.
| Species | Species Annotation Confidence | Ribosomal Nucleotide Identity | Nucleotide Overlap | Matching Library Alleles | Matching Library Nucleotides | PubMLST Multi-species DB Isolate ID | Ribosomal Nucleotide Identity Thresholds (A|B) |
|---|---|---|---|---|---|---|---|
| Klebsiella aerogenes | High | 99.947 | 100.000 | 51/51 | 20862/20862 | 55531 | 99.861|98.169 |
| Klebsiella variicola | Low | 98.111 | 99.990 | 51/51 | 20860/20862 | 123691 | 99.909|98.735 |
| Klebsiella quasivariicola | Low | 97.546 | 99.981 | 51/51 | 20858/20862 | 271266 | 99.94|99.736 |
| Klebsiella quasipneumoniae | Low | 97.531 | 99.990 | 51/51 | 20860/20862 | 173839 | 99.847|99.722 |
| Klebsiella africana | Low | 97.493 | 99.990 | 51/51 | 20860/20862 | 372794 | 99.950|99.741 |
Glossary
Alleles
Variant or alternative forms of a given gene. Alleles refer to sequence variation caused by different mutations in the same chromosomal location (Elston et al., 2012).
Antimicrobial resistance
Antimicrobial resistance occurs when antimicrobial drugs are used to treat infections caused by microorganisms such as bacteria, fungi, viruses and parasites. These drugs cause changes in the microorganism and allow antimicrobial resistance to develop, therefore making the antimicrobial that was originally effective, now ineffective at treating the infection (WHO, 2020).
Average nucleotide identity
The Average Nucleotide Identity (ANI) is a similarity index or measure of genomic similarity, at the nucleotide level, between the coding regions of two genomes. A cut-off score of >95% similarity between a pair of genomes indicates that they usually both belong to the same species (Arahal, 2014).
Concatenated
Concatenation assumes that individual genes in question have evolved along a common phylogeny. In a species tree, individual genes are linked together in a series to obtain a concatenated gene alignment. This method is more accurate than approaches that infer phylogenies using each gene separately, since a consensus of these gene phylogenies are used to generate a species tree.
ENA SRA
The European Nucleotide Archive (ENA) is a repository for annotated DNA sequences that also stores information about sequence assembly and metadata. The ENA consists of three databases, one of which is the Sequence Read Archive (SRA). The SRA stores DNA sequences generated through sequencing (Cochrane et al., 2008).
Genes/loci
A functional nucleotide sequence that encodes the synthesis of a protein (Raleigh, 2013).
Genotype
The genetic make-up of a microorganism identified by sequencing it’s DNA. The genotype of a microorganism can be used to predict phenotype.
Genus
A genus is a taxonomic rank used in the classification of living and fossil organisms. Genus sits between family and species (Baron, 1996).
MALDI-TOF
Matrix-assisted laser desorption/ionization (MALDI) is an ionization technique used in mass spectrometry that uses a laser energy absorbing matrix to create ions from a given biological sample. Time of flight (TOF) is a type of mass spectrometer that is widely used a capable of capturing a wide range of fragment masses. MALDI-TOF machines can be used to identify the species of bacteria present in a sample (Schumann and Maier, 2014).
Multilocus sequence typing (MLST)
DNA sequences (approx. 500 bp) from a set of housekeeping genes (approx. 7- 11) are used to characterise isolates within a species. For each housekeeping gene, the different sequences are assigned a distinct allele number. This is combined to generate an allelic profile or sequence type (ST) for each isolate, allowing isolates within the same species to be compared (Maiden et al., 1998).
NCBI database
The National Center for Biotechnology Information (NCBI) provides analysis and retrieval of resources from multiple databases. The NCBI database also houses the GenBank database for public DNA sequences, and the Taxonomy database which includes curated classification and nomenclature for DNA sequences (NCBI Resource Coordinators, 2012).
Neighbour joining tree
A neighbour joining tree is a clustering method used to generate phylogenetic trees. The clustering algorithm uses information from DNA sequence data to generate a tree. At each stage of clustering, the total branch length is minimised by finding pairs of DNA sequences between isolates (Saitou and Nei, 1987).
Paralogous
Genes that occur multiple times in the genome, as a result of gene duplication.
Phenotype
Phenotypes are observable characteristics of cells that can be identified by studying the morphology and antibiotic susceptibility of a microorganism (Bochner, 2009).
Ribosomal DNA
The DNA that codes for the ribosomal RNA. The gene coding for the prokaryotic 16S ribosomal subunit is highly conserved between different species and can therefore be used to study phylogenetic relationships (Kolbert and Persing, 1999).
Ribosomal MLST
Please refer to MLST first. Ribosomal MLST uses MLST to index variation specifically among the 53 bacterial ribosome subunit (rps) genes (Jolley et al., 2012).
Scheme
A collection of loci with a common association that allows them to be grouped together.
Species
A bacterial species is considered to be “a monophyletic and genomically coherent cluster of individual organisms that show a high degree of overall similarity with respect to many independent characteristics, and is diagnosable by a discriminative phenotypic property” (Rosselló-Mora and Amann, 2001)
A good description is given here: https://help.ezbiocloud.net/bacterial-species-concept-explained/
Type strains
The first strain of a species isolated, when the species was first described in a publication (Public Health England, 2020).
16S rRNA sequencing
The bacterial ribosome consists of 3 rRNA molecules and is important for translating messenger RNA into proteins. The 16S ribosome is the central structural component of the bacterial and archaeal 30S ribosomal subunit and is required for the initiation of protein synthesis. The rRNA genes, especially those for 16S rRNA form the structural core of the ribosome and are least likely to experience horizontal gene transfer between species. For this reason, 16s rRNA sequencing is used to identify variation within a genus or species (Barghouthi, 2011).
Conserved regions of the 16S rRNA gene allow the design of universal primers that bind to and amplify the chromosomal target region. A molecular biology method called PCR (polymerase chain reaction) allows amplification of these specific DNA targeted regions. This is followed by DNA sequencing (Bukowska-Faniband et al., 2020).
References
Genomic methods used to validate species
Lee I, Ouk Kim Y, Park SC, Chun J. OrthoANI: An improved algorithm and software for calculating average nucleotide identity. Int J Syst Evol Microbiol. 2016;66(2):1100-3.
Ondov BD, Treangen TJ, Melsted P, Mallonee AB, Bergman NH, Koren S, et al. Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016;17(1):132.
Wick RR, Heinz E, Holt KE, Wyres KL. Kaptive Web: User-Friendly Capsule and Lipopolysaccharide Serotype Prediction for Klebsiella Genomes. Journal of clinical microbiology. 2018;56(6):e00197-18.
What is rMLST
Jolley KA, Bliss CM, Bennett JS, Bratcher HB, Brehony C, Colles FM, et al. Ribosomal multilocus sequence typing: universal characterization of bacteria from domain to strain. Microbiology (Reading). 2012;158(Pt 4):1005-15.
Timeline
Barrios-Camacho H, Aguilar-Vera A, Beltran-Rojel M, Aguilar-Vera E, Duran-Bedolla J, Rodriguez-Medina N, et al. Molecular epidemiology of Klebsiella variicola obtained from different sources. Scientific Reports. 2019;9(1):10610.
Brisse S, Passet V, Grimont PAD. Description of Klebsiella quasipneumoniae sp. nov., isolated from human infections, with two subspecies, Klebsiella quasipneumoniae subsp. quasipneumoniae subsp. nov. and Klebsiella quasipneumoniae subsp. similipneumoniae subsp. nov., and demonstration that Klebsiella singaporensis is a junior heterotypic synonym of Klebsiella variicola. International Journal of Systematic and Evolutionary Microbiology. 2014;64(Pt_9):3146-52.
Dworkin M, Falkow S, Rosenberg E, Schleifer KH, Stackebrandt E. The Prokaryotes: Vol. 6: Proteobacteria: Gamma Subclass: Springer New York; 2006.
Long SW, Linson SE, Ojeda Saavedra M, Cantu C, Davis JJ, Brettin T, et al. Whole-Genome Sequencing of a Human Clinical Isolate of the Novel Species Klebsiella quasivariicola sp. nov. Genome announcements. 2017;5(42):e01057-17.
Merla C, Rodrigues C, Passet V, Corbella M, Thorpe HA, Kallonen TVS, et al. Description of Klebsiella spallanzanii sp. nov. and of Klebsiella pasteurii sp. nov. Frontiers in microbiology. 2019;10:2360-.
Rodrigues C, Passet V, Rakotondrasoa A, Diallo TA, Criscuolo A, Brisse S. Description of Klebsiella africanensis sp. nov., Klebsiella variicola subsp. tropicalensis subsp. nov. and Klebsiella variicola subsp. variicola subsp. nov. Res Microbiol. 2019;170(3):165-70.
Rosenblueth M, Martínez L, Silva J, Martínez-Romero E. Klebsiella variicola, a novel species with clinical and plant-associated isolates. Syst Appl Microbiol. 2004;27(1):27-35.
Wyres KL, Lam MMC, Holt KE. Population genomics of Klebsiella pneumoniae. Nature Reviews Microbiology. 2020;18(6):344-59.
Glossary
Arahal DR. Chapter 6 - Whole-Genome Analyses: Average Nucleotide Identity. In: Goodfellow M, Sutcliffe I, Chun J, editors. Methods in Microbiology. 41: Academic Press; 2014. p. 103-22.
Barghouthi SA. A universal method for the identification of bacteria based on general PCR primers. Indian journal of microbiology. 2011;51(4):430-44.
Baron EJ. Classification. In: Baron S, editor. Medical Microbiology. 4th edition. Galveston (TX): University of Texas Medical Branch at Galveston; 1996. Chapter 3.
Bochner BR. Global phenotypic characterization of bacteria. FEMS Microbiol Rev. 2009;33(1):191-205.
Bukowska-Faniband EA, Tilde. Lood, Rolf. 16S rRNA Sequencing: A PCR-based Technique to Identify Bacterial Species | Protocol. JOVE. 2020.
Cochrane G, Akhtar R, Aldebert P, Althorpe N, Baldwin A, Bates K, et al. Priorities for nucleotide trace, sequence and annotation data capture at the Ensembl Trace Archive and the EMBL Nucleotide Sequence Database. Nucleic acids research. 2008;36(Database issue):D5-D12.
Elston RC, Satagopan JM, Sun S. Genetic terminology. Methods in molecular biology (Clifton, NJ). 2012;850:1-9.
Jolley KA, Bliss CM, Bennett JS, Bratcher HB, Brehony C, Colles FM, et al. Ribosomal multilocus sequence typing: universal characterization of bacteria from domain to strain. Microbiology (Reading, England). 2012;158(Pt 4):1005-15.
Kolbert CP, Persing DH. Ribosomal DNA sequencing as a tool for identification of bacterial pathogens. Current Opinion in Microbiology. 1999;2(3):299-305.
Maiden MCJ, Bygraves JA, Feil E, Morelli G, Russell JE, Urwin R, et al. Multilocus sequence typing: A portable approach to the identification of clones within populations of pathogenic microorganisms. Proceedings of the National Academy of Sciences. 1998;95(6):3140.
NCBI Resource Coordinators. Database resources of the National Center for Biotechnology Information. Nucleic Acids Research. 2012;41(D1):D8-D20.
Public Health England. Type Strains – the most important strains in NCTC? : Culture Collections; 2020 [Available from: https://www.phe-culturecollections.org.uk/news/nctc-news/type-strains-%E2%80%93-the-most-important-strains-in-nctc.aspx]
Raleigh EA. Bacterial Genes. In: Maloy S, Hughes K, editors. Brenner's Encyclopedia of Genetics (Second Edition). San Diego: Academic Press; 2013. p. 262-4.
Rosselló-Mora R, Amann R. The species concept for prokaryotes. FEMS Microbiology Reviews. 2001;25(1):39-67.
Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4(4):406-25.
Schumann P and Maier T, MALDI-TOF mass spectrometry applied to classification and identification of bacteria, Methods in Microbiology, 41 (2014), pp. 275-306
World Health Organisation. Antimicrobial resistance 2020 [Available from: https://www.who.int/news-room/fact-sheets/detail/antimicrobial-resistance].